de | fr | en print view![]()
3R-INFO-BULLETIN 29
May 2005
The Authors

Prof. Angelo Vedani (middle) is Director of the Biographics Laboratory 3R in Basel and Associate Professor at the Department of Pharmacy, University of Basel, Switzerland. Prof. Max Dobler (left) is Professor emeritus, ETH Zürich and a trained computational scientist. Dr. Markus Lill (right) currently directs ADMET activities at the Biographics Laboratory 3R and is completing his habilitation at the University of Basel.
Current addresses:
Angelo Vedani
admin@biograf.ch
Biographics Laboratory 3R
Friedensgasse 35
CH-4055 Basel
Switzerland
www.biograf.ch
Editor
Peter Maier, Scientific Adviser of the 3R Research Foundation
Computer-based quantification of (adverse) effects triggered by drugs and chemicals
Reception of chemicals at biological structures
The Interaction with receptors plays an important role in drug discovery (Computer-Aided Drug-Discovery, CADD) and in the assessment of the potential interaction of new or existing chemicals (e.g. REACH). At the Biographics Laboratory 3R in Basel, models for nuclear receptors (→ endocrine disruption) and for cytochrome P450 3A4 (→ metabolic transformations) were developed.[*] For the toxicologically relevant aryl hydrocarbon receptor (AhR), this includes the establishment of three-dimensional surrogates for a binding pocket (virtual receptor) in absence of the experimental structure.
Design of (virtual) receptor models
The process is based on a receptor-modeling concept which combines flexible docking (software Yeti) and multidimensional QSAR (software Quasar1). Quasar generates a family of quasi-atomistic receptor-surface models that are optimized by means of a genetic algorithm. The fourth dimension (→ 4D-QSAR) refers to the possibility of representing each ligand molecule as an ensemble of conformations, orientations and protonation states, thereby reducing the bias in identifying the bioactive conformer. The fifth dimension (→ 5D-QSAR) allows to evaluate different induced-fit protocols simultaneously while the sixth dimension (→ 6D-QSAR) allows to consider different solvation models. A different philosophy (dual-shell representation of the binding site) has been incorporated in the software Raptor2 which - in combination with Quasar - allows for consensus scoring.
Nuclear receptors
Nuclear receptors comprise a family of ligand-dependent transcription factors that transform extra- and intracellular signals into cellular responses by triggering the transcription of target genes. In particular, they mediate the effects of hormones (ligand, e.g. the estrogens (ER), androgens (AR), progesterones, glucocorticoids) and hormonally active compounds (endocrine disruptors). A number of receptor-mediated adverse effects by xenobiotics have been identified in the past. This includes toxicity mediated by the thyroid hormone receptor, the epidermal growth factor and aryl hydrocarbon receptor (AhR). The concern about chemicals which bind to these receptors and induce adverse, uncontrolled effects has created a need to both screen and monitor compounds before they are manufactured, used or released into our environment. For the ER and AR, the binding mode was identified by using the X-ray crystal structure as a template.3 As the protein can accommodate different binding modes with only moderate rearrangement of side chains located in the active-site pocket (→ induced fit), it is necessary to sample all energetically feasible modes and compose them into a 4D data set. For the AhR no experimental structure is available. Here, the ligand superposition was accomplished based on the molecular skeleton4.
Validation of receptors
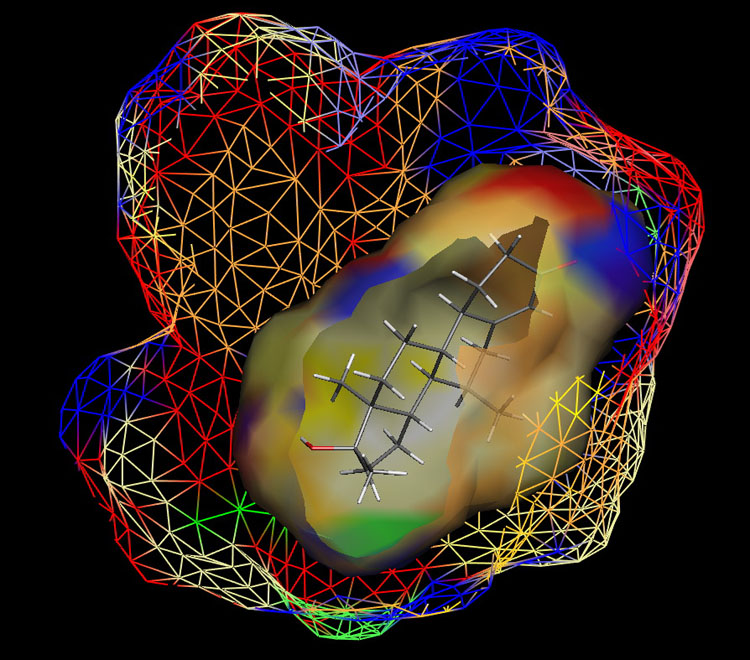
Fig. 1: Raptor model of the AR.
The ER study was based on 106 compounds (six substance classes, 88 training, 18 test ligands, 0.2 nM ≤IC50 ≤ 2.8 mM), the AR simulation on 119 compounds (6/88/31, 1.6 nM ≤ IC50 ≤ 0.5 mM) and the AhR analysis on 121 compounds (4/91/30, 1.0 nM ≤ IC50 ≤ 3.2 mM). Using the Quasar (ER, AhR) and Raptor (AR) technologies (Figure 1), the simulations converged at cross-validated r2 (correlation coefficient) ranging from 0.832 to 0.903 and yielded predictive r2 of 0.727, 0.885 (Figure 2a) and 0.754 (Figure 2b) . Most importantly, we could show5 that our receptor surrogates are able to correctly predict the binding affinity of compounds different from those in the training set (AR: Figure 2b: yellow+green dots and http://www.biograf.ch/projects.html).
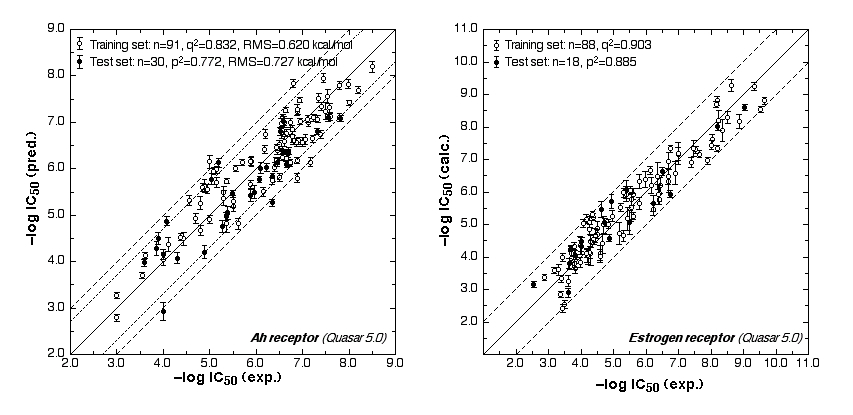
Fig. 2a: Comparison of calculated and experimental IC50 values (left panel: AhR, right panel: ER).
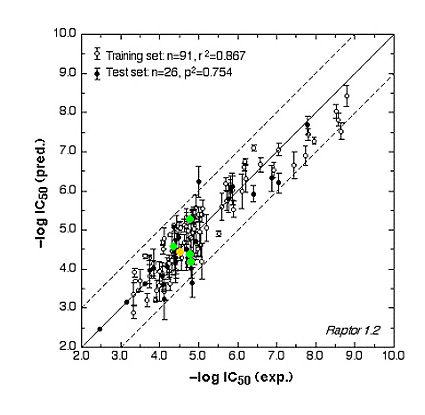
Fig. 2b: Comparison of calculated and experimental IC50 values of the AR receptor.
Metabolic transformation
Competition of drugs for metabolization at Cytochrome P450 3A4 (CYP3A4) may result in undesired drug-drug interactions. In addition, CYP3A4 might transform chemicals into reactive metabolites. The development of a computational model to accurately predict the docking potential of a diverse set of ligand molecules was based on the X-ray crystal structure of the human CYP3A4 enzyme (→ Figure 3a, → flexible docking) and a total of 48 structurally diverse molecules (38 training and 10 test compounds). The simulation reached a cross-validated r2 of 0.888 and yielded a predictive r2 of 0.746 (Figure 3b).
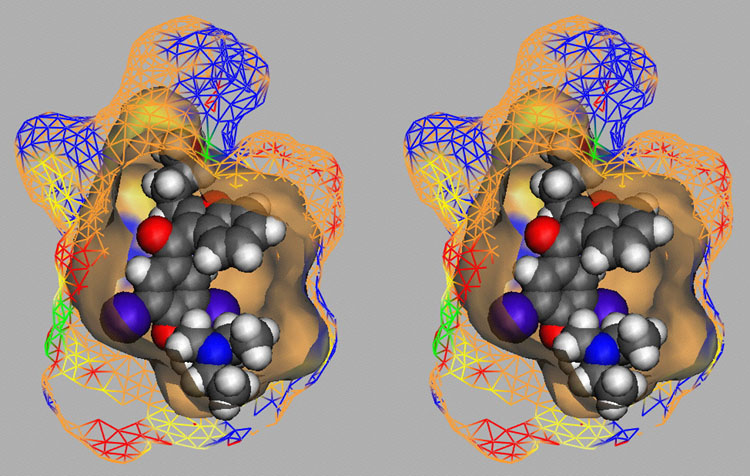
Fig. 3a: Raptor model of the CYP3A4 binding pocket.
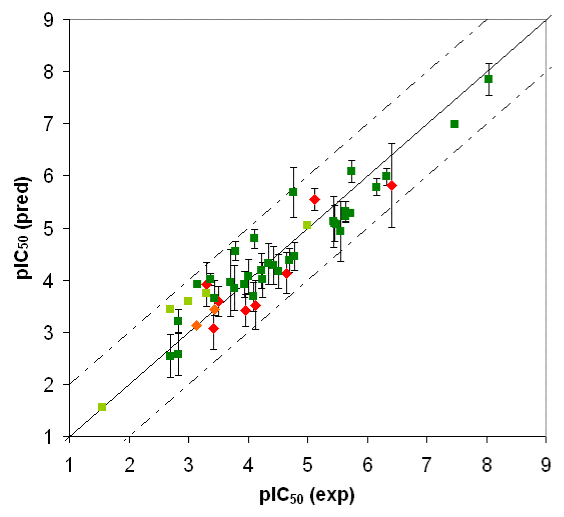
Fig. 3b: Comparison of calculated and experimental IC50 values of CYP3A4. Dark green = training set; red = test set. Threshold compounds: light green = training set; orange = test set.
Testing via Internet
The Biographics Laboratory 3R is planning to implement a database on the Internet for the screening of adverse effects triggered by drugs and chemicals in silico. The systems described in this account (AhR, ER, AR, CYP3A4) represent its backbone; other entities include the 5HT2A, the cannabinoid and GABAA receptor. Within this framework, new - hypothetical or existing - compounds can now be tested for their activity towards the various receptors (models) and their potential harmfulness may be deducted therefrom. Toxicity or adverse effects mediated by receptors other than those stored in the database will not be identified. Accordingly, the present approach based on receptor modeling will result in the production of false-negative results for classes of toxic chemicals which do not interact via receptor or which interact via so far unknown receptor-based pathways. Therefore, QSAR technologies may be used to identify the harmful potential of a drug or chemical and no false positives are produced. However, they are not suited to prove its harmlessness.
3R Relevance?
Such in silico techniques are considered as valuable alternatives to in vivo or in vitro screening approaches. The described technology enables the identification of harmful drugs or chemicals long before in vivo experiments are typically conducted. In drug development, identified substances will not be further elaborated. Molecules with more potential would take their place and animal tests will be limited to drugs with less toxic effects and subsequently higher chances for further development. Within the REACH framework, the number of false-negative results may be reduced by expanding the database with additional receptor models. Identified positive chemicals could be sent, without further animal tests, to risk assessement. Both concepts substantially reduce animal testing.
Published updated version of this Bulletin 29/2007 (PDF)
References:
- Vedani, A. and Dobler, M. 5D-QSAR: The key for simulating induced fit? J. Med. Chem. 2002, 45, 2139–2149.
- Lill, M.A., Vedani, A. and Dobler, M. Raptor - combining dual-shell representation, induced-fit simulation and hydrophobicity scoring in receptor modeling: Application towards the simulation of structurally diverse ligand sets. J. Med. Chem. 2004 ,47, 6174–6186.
- Vedani, A. and Huhta, D.W. A new force field for modeling metalloproteins. J. Am. Chem. Soc. 1990, 112, 4759-4767.
- Vedani, A., Dobler, M. and Lill, M.A. In silico prediction of harmful effects triggered by drugs and chemicals. Toxicol. Appl. Pharmacol. 2005 (in press).
- Lill, M.A., Dobler, M. and Vedani, A. In silico prediction of receptor-mediated environmental toxic phenomena — Application towards endocrine disruption. SAR QSAR Environ. Res. 2005 ,16, 149–169.
| [*] | Interaction between a chemical and a particular receptor can be foreseen in silico.Computer-based (in silico) concepts have matured into powerful tools for simulating and quantifying biochemical processes at the molecular level. This has become possible by a wide body of protein structures (cf. http://www.rcsb.org/pdb) available at atomic resolution, a detailed understanding of the forces governing molecular interactions and the leap in computing power. The computer-aided simulation of a possible specific interaction between a chemical and a biological structures is based on the three-dimensional complementarity of substrate and receptor, recognized in 1894 by Emil Fischer (→ lock-and-key analogon; Nobel prize in 1902). Nowadays, the technology allows to identify stereospecific and selective activity of a given molecule. In drug development, this can be used by tailor-making their properties (scaffold, functional groups) directly at the binding pocket of the true biological receptor (→ structure - based design). Unfortunately, true (experimental) structures are lacking for a majority of receptors of biomedical interest. The Biographics Laboratory 3R has pioneered computational concepts allowing to construct a three-dimensional surrogate for a binding pocket (virtual receptor) in absence of the experimental structure (→ receptor modeling). Such binding-site models are validated by establishing a Quantitative Structure-Activity Relationship (QSAR) for a series of compounds with known interaction at the defined biological relevant receptor (with unknown structure) for training and the actual test compounds with unknown activity. |
| Last modified 2008/01/30 |