Angelo Vedani
Biographics Laboratory 3R, 4056 Basel, Switzerland
present address: Department of Chemistry, Molecular Modeling, University of Basel
Pharmacenter, 4056 Basel, Switzerland
angelo.vedani@unibas.ch
Keywords: drug-design; pharmacology; qsar; in silico; reduction; replacement; toxicity testing: pharmaceuticals; toxicity testing: receptor mediated; toxicity testing: xenobiotics
Duration: 1 year Project Completion: 2003
Background and Aim
We have created three-dimensional surrogatevirtual models of cellular receptors involved in chemically induced toxicity. The simulation uses a 4D/5D-QSAR concept developed at our laboratory (Vedani et al. 2000; Vedani and Dobler 2000). This model allows various chemical details and technical aspects of receptor – ligand interactions to be examined: multiple ligand presentation, induced fit, H-bond flip-flop and dynamic cavity shaping. As an example, we created a virtual Aryl hydrocarbon receptor (Ah receptor; this receptor binds dibenzodioxins, dibenzofurans, biphenyls, and polyaromatic hydrocarbons), and used a total of 121 binding compounds to calibrate the model and examine its behaviour. Our results yielded a cross-validated r2 of 0.851 and a predictive r2 of 0.792 (Fig.1). None of the compounds in either the training or the test set predicted false positive or false negative results (Vedani and Dobler 2001).
It is our objective to establish a virtual laboratory on the Internet to allow the harmful effects triggered by drugs, chemicals and their metabolites to be estimated in a virtual environment.
Method and Results
Free access to this virtual laboratory will allow any interested party to estimate the toxic potential of a given substance, even before the compound has been synthesised! This will be done by first modelling the three-dimensional structure of the substance and its possible metabolites in the computer, and then calculating the binding affinity of these compounds towards each receptor modelled in the database.
Testing a new compound for harmful effects involves the following steps: 1. Downloading the 2D/3D data of the substance on our server (or a local mirror) via an Internet protocol. 2. Automated generation of the structures of potential metabolites. 3. Identifying the most likely (i.e energetically favourable) 3D structural conformations for the parent compound and the metabolites. 4. Identifying likely positions and orientations of the parent compound and metabolites in interaction with every receptor in the database (using a Monte-Carlo search procedure). 5. Estimating the harmful potential of a substance by calculating its binding affinity and that of all metabolites towards each receptor.
Only those compounds and metabolites that pass through this virtual screening test with binding constants weaker than a threshold value (e.g. K> 50 mM) may be considered “safe” and cleared for further studies, including animal tests.
Presently, our database includes validated models for the Ah receptor (receptor-mediated toxicity), the 5HT-2A receptor (hallucinogenic activity), the cannabinoid receptor (psychotropic effects), the GABA-A receptor (receptor-mediated toxicity), and the steroid receptor (various undesired effects). The database will be continuously extended to include models of further receptors known or presumed to mediate toxicity or other harmful effects (Fig.2).
The following steps are necessary to add a new receptor to the database: 1. Selecting the training and test sets of ligands for which experimental binding data is available. 2. Generating all 3-D structures. 3. Conformational search. 4. Generating and validating the new receptor model. 5. Adding the model into the data base and external testing.
A noteworthy feature of this system is that the classified or proprietary data is used only for model construction but not for testing and validation, i.e. contributions from the pharmaceutical industry can be generated at their laboratories using a local mirror of the database.
Conclusions and Relevance for 3R
The proposed Internet laboratory contributes to two aspects of the 3R principle: Firstly, it allows potentially harmful substances, e.g. potential drug candidates, to be recognised in an early phase of development, before the compounds are synthesised or tested in preclinical trials including animal tests. In the case of industrial chemicals, e.g. chemicals that entered the EU market before 1981, the database will also contribute to the identification of harmful substances already on the market. Secondly, the availability of the database should help to prevent redundancies in the testing of potential drugs or chemicals with identical or closely related biomedical targets.
(see also 3R-INFO-BULLETIN Nr. 29)
Published updated Version 29/2007 (pdf)
References
1. Vedani, A. and Dobler, M. (2000). Multidimensional QSAR in drug research: Predicting binding
affinities, toxicity, and pharmacokinetic parameters. Progress in Drug Research, Jucker, E. (ed.),
Birkhäuser, Basel/Boston/Berlin, 55, 105-135.
2. Vedani, A. and Dobler M. (2001). Internet laboratory for predicting harmful effects triggered by drugs
and chemicals. Concept and call for co-operation. ALTEX, 18, 110-114.
3. Vedani, A. and Dobler, M. 5D-QSAR: The key for simulating induced fit? J. Med. Chem. 2002, 45, 2139–2149.
4. Lill, M.A., Vedani, A. and Dobler, M. Raptor - combining dual-shell representation, induced-fit simulation and hydrophobicity scoring in receptor modeling: Application towards the simulation of structurally diverse ligand sets. J. Med. Chem. 2004 ,47, 6174–6186.
5. Vedani, A. and Huhta, D.W. A new force field for modeling metalloproteins. J. Am. Chem. Soc. 1990, 112, 4759-4767
6. Vedani, A., Dobler, M. and Lill, M.A. In silico prediction of harmful effects triggered by drugs and chemicals. Toxicol. Appl. Pharmacol. 2005 (in press).
7. Lill, M.A., Dobler, M. and Vedani, A. In silico prediction of receptor-mediated environmental toxic phenomena — Application towards endocrine disruption. SAR QSAR Environ. Res. 2005 ,16, 149–169.
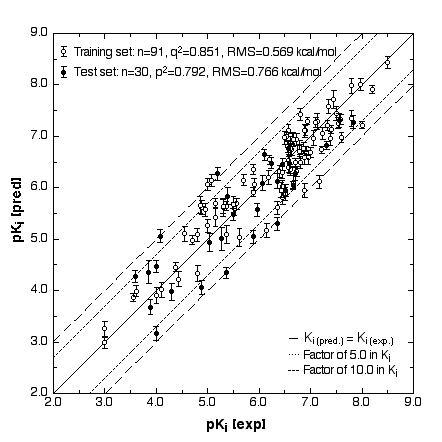
Figure 1
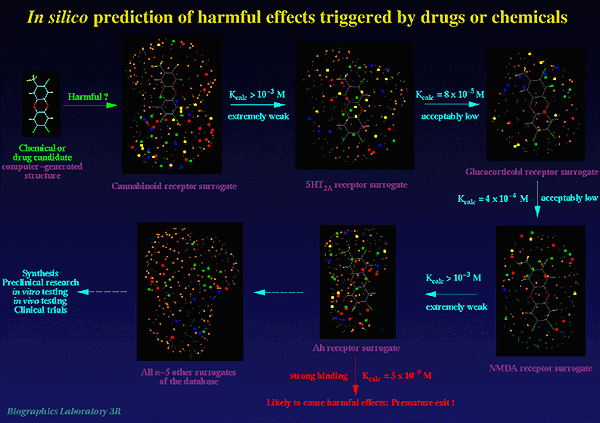
Figure 2