Matthias P. Lutolf
Institute of Bioengineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
matthias.lutolf@epfl.ch
Keywords: human; mice; stem cells; transplantation; reduction; replacement
Duration: 1 year Project Completion: 2013
Background and Aim
Although hematopoietic stem cells (HSC) are the most successfully used clinical stem cells (1), their transplantation to treat hematological malignancies remains a risky procedure with major complications including infections and graft-versus-host disease. Moreover, the limited availability of human leukocyte antigen (HLA)-matched donors and the low numbers of transplantable HSCs that can be isolated from a donor are major issues that could potentially be overcome if efficient in vitro HSC expansion methods were available. Yet, as soon as HSCs are removed from their native microenvironmental niches in the bone marrow and put in standard cell culture, they lose their characteristic functions by undergoing rapid differentiation.
Progress to discover robust in vitro systems capable to expand HSCs has been hampered by a lack of prospective markers that predict reconstitution activity of cultured HSCs. Indeed, the only functional assay to probe long-term HSC multipotency remains transplantation, a method that is time-consuming, entails animal suffering and thus raises ethical concerns. Classical in vitro assays to assess hematopoiesis such as colony formation in a semi-solid matrix that contains cytokines are a powerful means to assess clonogenicity and differentiation potential of an unknown population of stem/progenitor cells. However, such assays are retrospective and fail to predict long-term reconstitution activity. Furthermore, it is well accepted in the field that immunostaining in vitro-cultured HSCs does not faithfully mark functional stem cells.
Given these problems, the main goal of this 1-year project is to identify predictive metabolic markers of the HSC state and to validate these markers on stem cells exposed in vitro to self-renewal and differentiation culture conditions, as well as different oxygen tensions. In contrast to the widely used ‘immunophenotyping’ of stem and progenitor cell populations via antibodies against specific receptors, we will focus on metabolic markers of the stem/progenitor cell state, since recent studies have shown that HSC quiescence and self-renewal are at least partially controlled through regulation of their metabolic state (e.g. (2)).
Method and Results
in progress (present status)
Long-term HSCs in mice are exclusively marked by low metabolic activity (preliminary data)
In a preliminary experiment, we tested whether progenitor/stem cells having lower metabolic activity represent long-term repopulating (i.e. functional) HSCs. To this end, Lineagenegative Sca-1positive c-kitpositive (LKS) cells were purified by FACS based on mitochondria activity using tetramethylrhodamine methyl ester (TMRM), a cell-permeant fluorescent dye that is sequestered by active mitochondria (Figure 1).
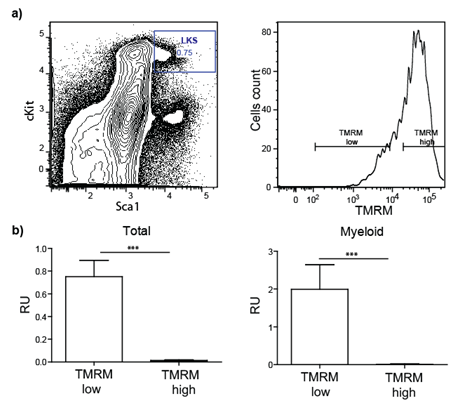
Figure 1: Transplantation of sorted stem/progenitor cells based on mitochondria activity. a) LKS are sorted in two populations based on their TMRM intensity (low and high). b) Total blood and myeloid compartment analysis of lethally irradiated transplanted mice demonstrate that only cells TMRM low (low mitochondrial activity) engraft successfully.
Probing mitochondria activity by microscopy-based techniques
Based on these encouraging data, we aim to establish a microscopy-based method to analyze and distinguish mitochondria in primary HSCs and progenitors in vitro. First, we will stain mitochondria by mitotracker and TMRM in freshly isolated, fixed HSC/MPPs and analyze them by confocal microscopy at high magnification. One read-out that we plan to use is the mitochondrial morphology that can be indicative of the mitochondrial activation status of stem cells (3). Mitochondria with low metabolic activity can thus be expected to have a punctated morphology, whereas highly metabolic mitochondria tend to fuse and form a network-like structure (4). We will develop image analysis tools to quantify individual mitochondria in single cells in order to extract information on the stemness. Furthermore, a measurement of the fluorescence intensity of the TMRM staining should provide a good read-out of mitochondrial activity. Finally, we will analyze mitochondrial morphology (by Mitotracker) and activity (by TMRM) in live single cells by fluorescent (confocal) microscopy, which could yield a potent in vitro read-out on the stem versus progenitor cell state.
Assessment of metabolic marker changes during in vitro culture
We will apply the above-mentioned read-outs to monitor how individual HSCs change their metabolic phenotype during in vitro culture conditions, inducing known self-renewal versus differentiation divisions, as well as under hypoxic culture conditions (1-5%, using premixed gas with defined oxygen composition). As the native HSC niche is an extremely hypoxic microenvironment that forces cells into low metabolism, we expect hypoxic conditions to contribute to the maintenance of the stem cell state in vitro as well. Oxygen tension will be measured using an oxygen-sensitive ruthenium probe (FOXY-slide, Ocean Optics, Inc.).
Conclusions and Relevance for 3R
HSC transplantations, currently the only functional means to identify and characterize mammalian HSCs, are radical and expensive procedures used in countless labs all over the world. Therefore, the identification of novel in vitro markers that are predictive of the functional HSC state would be highly significant. Among other things, it could open up new avenues to rapidly identify stem cells under conditions of in vitro expansion.
References
1. D. Bryder, D. J. Rossi, I. L. Weissman, Am J Pathol 169, 338 (Aug 1, 2006).
2. T. Suda, K. Takubo, G. L. Semenza, Cell Stem Cell 4, 298 (2011).
3. A. Prigione, B. Fauler, R. Lurz, H. Lehrach, J. Adjaye, Stem Cells 28, 721 (Apr, 2010).
4. B. Westermann, Embo Rep 3, 527 (Jun, 2002).